UDK: 616-056.7-089.163-053.2
Mikjunovikj – Derebanova Lj1, Kuzmanovska B1, Donev Lj1, Lleshi A1, Demjanski V1, Cvetanovska – Naunova V2
1 University Clinic for Anesthesia, Reanimation and Intensive Care Medicine, University “Ss. Cyril and Methodius”, Skopje, Republic of North Macedonia
2 University Clinic for Pediatric Surgery, University “Ss. Cyril and Methodius”, Skopje, Republic of North Macedonia
Abstract
Introduction
Kabuki Syndrome is a rare congenital disease with characteristic phenotypic appearance that includes arched eyebrows with sparseness of the lateral one – third, long palpebral fissures, eversion of the lateral third of the lower eyelid, a short columella with a depressed nasal tip and prominent ears. Other comorbid conditions include seizers and hypotonia, growth impairment, congenital heart diseases, endocrine involvement and renal abnormalities, as well as developmental delay and intellectual disability. The aim of this case was to describe the perioperative management of a child with Kabuki Syndrome and to discuss the potential problems in anesthesia.
Case Presentation
An 11 months old male child was admitted to the Clinic for Pediatric Surgery for elective operation for right-sided inguinal hernia. The patient was with growth delay (weighted 4.5kg), with hypotonia, without the ability to sit independently, nor to stand upright. Occasionally he was able to control his head movement, but he could not follow with his eyes. Echocardiography showed ASD secundum with L – D shunt without hemodynamic reflection. His antiepileptic therapy was canceled one moth earlier by his pediatric neurologist. In the physical examination, significantly long palpebral fissures and thinness in the 1/3 lateral of the highly curved eyebrows were observed. He had eversion in his lower eyelids. His nasal septum was low, and his ears were low set. The induction was started with 0.5mg/kg lidocaine, 1mcg/kg fentanyl and 4mg/kg propofol. Mask ventilation was easily performed and 0.6mg/kg rocuronium was given intravenously. The patient was intubated with a 3.0 cuffed tube using video laryngoscope. Caudal block was performed after the induction with 0.5ml/kg bupivacaine 0.25%. The anesthesia was maintained with 4-6 mg/kg/hour propofol. No complications were reported in the postoperative period.
Conclusion
In perioperative management of the patient with Kabuki Syndrome, anesthesiologist should take under consideration the possible difficult intubation, neurological (seizure) and musculoskeletal (hypotonia) disorders, cardiac (CaA, ASD, VSD) abnormalities, respiratory problems (recurrent infections), urogenital (hydronephrosis, renal hypoplasia) abnormalities and a latex allergy and the risk of malignant hyperthermia.
Key Words: caudal block, general anesthesia, Kabuki Syndrome, pediatric anesthesia, perioperative management.
Introduction
Kabuki Syndrome is a rare congenital disease that occurs with an incidence of 1:32000 live births in Japan and 1:80000 in the rest of the world (1). The patients’ facial appearance with this syndrome resemble to the traditional make-up used in Japanese theater Kabuki and the term Kabuki make-up syndrome was suggested by Niikawa (2,3). Patients with Kabuki Syndrome have characteristic phenotypic appearance that include arched eyebrows with sparseness of the lateral one – third, long palpebral fissures, eversion of the lateral third of the lower eyelid, a short columella with a depressed nasal tip and prominent ears (Figure 1). Also, patients with this syndrome can have a wide range of clinical presentation from deformities of the skeletal-muscular system, high palate, dental anomalies, short stature, cardiovascular anomalies, renal malformations, frequent pneumonias, otitis media and hearing loss. During surgical interventions, attention should be paid also to concomitant congenital anomalies.
The aim of this case was to describe the perioperative management of a child with Kabuki Syndrome and to discuss the potential problems in anesthesia.

Figure 1. Characteristic phenotype appearance of patient with Kabuki Syndrome (This picture is taken from Boniel S, Szymańska K, Śmigiel R and Szczałuba K. Kabuki Syndrome – Clinical Review with Molecular Aspects. Genes 2021, 12(4), 468; https://doi.org/10.3390/genes12040468)
Case Presentation
An 11 months old male child was admitted to the Clinic for Pediatric Surgery for elective operation for right-sided inguinal hernia. According to the medical history received from the patient’s mother, the patient was born weighing 2,330gr at the end of the eighth month of pregnancy with C–section. The patient had been diagnosed with Kabuki Syndrome when he was 6 months old as a result of the genetic tests made due to neuromotor growth deficiency and dysmorphism. A genetic mutation in the KDM6A gene in a hemizygous state was proven in the patient. This gene is responsible for coding of histone demethylase protein. KDM6A-associated Kabuki Syndrome is characterized by typical facial dysmorphia, skeletal abnormalities, hypotonia, intellectual and developmental delays, hypoglycemic hyperinsulinism and increased susceptibility to infections. De novo mutations with X – linked dominant inheritance are the most often observed.
At birth, the patient was diagnosed with intraventricular hemorrhage and had been in incubator for 8 days. At the age of 4 months, the patient was hospitalized due to osteomyelitis of the right hip, and he was treated only with antibiotic therapy.
At the admission for elective inguinal hernioplasty, the patient was with growth delay (weighted 4.5kg), with hypotonia, without the ability to sit independently, nor to stand upright. Occasionally he was able to control his head movement, but he could not follow with his eyes. Hematological and biochemical investigations showed no abnormalities. Echocardiography showed ASD secundum with L – D shunt without hemodynamic reflection. His antiepileptic therapy was canceled one month earlier by his pediatric neurologist. In the physical examination, significantly long palpebral fissures and thinness in the 1/3 lateral of the highly curved eyebrows were observed. He had eversion in his lower eyelids (Figure 2). His nasal septum was low, and his ears were low set (Figure 3).
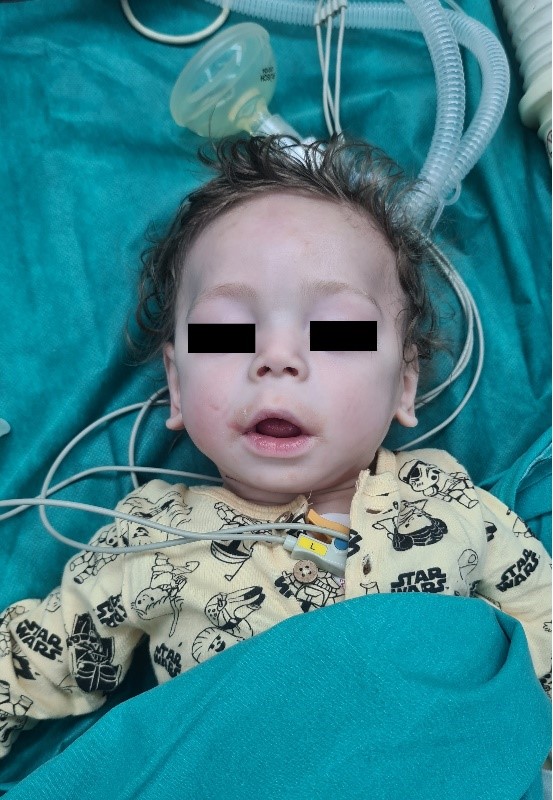
Figure 2. Long palpebral fissures and thinness in the 1/3 lateral of the highly curved eyebrows. But the columella in this case was not short.
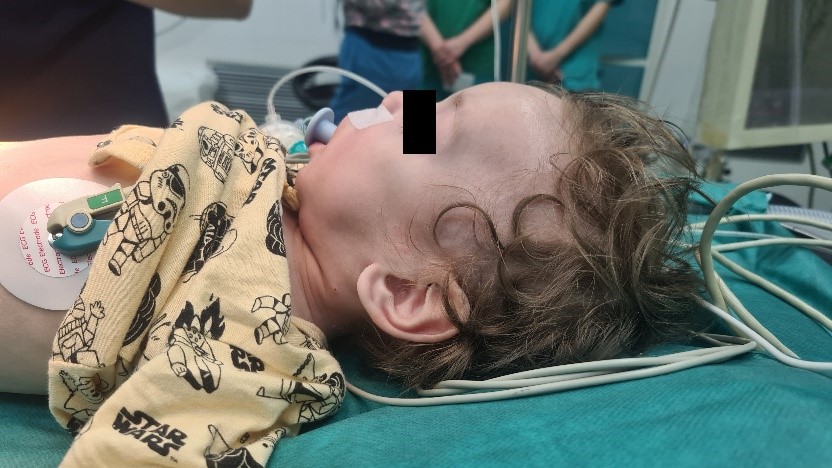
Figure 3. Low nasal septum, low – set prominent and mandibular hypoplasia
On the operation day, the patient was premedicated with 0.5mg/kg oral midazolam. Essential precautions were taken against the potentiality of difficult intubation. The patient was monitored, blood pressure was 108/70mmHg, heart rhythm was regular with rate 140 beats/minutes and oxygen saturation was 98% in the room air. Vascular access was secured, and the induction was started with 0.5mg/kg lidocaine, 1mcg/kg fentanyl and 4mg/kg propofol. After we were sure that we can ventilate the patient, 0.6mg/kg rocuronium was given intravenously, and the patient was intubated with a 3.0 cuffed tube using video laryngoscope. Caudal block was performed after the induction, and 0.5ml/kg of bupivacaine 0.25% was administrated. Anesthesia was maintained with 4-6mg/kg/hour propofol. The operation ended one hour after the induction, and the patient was awakened. No complications were reported in the postoperative period.
Discussion
Kabuki Make-up Syndrome or Niikawa-Kuroki Syndrome was first described in 1981 by the two Japanese physicians N. Niikawa (2) and Y. Kuroki (3). Kabuki Syndrome is a rare genetic disorder and results from de-novo mutations of the KMT2D gene on chromosome 12 (KMT2D – associated, autosomal – dominant KS type 1) or de-novo deletions of the KDM6A gene on the X – chromosome (KDM6A – associated, X – linked – dominant KS type 2) (4,5). Up to 75% of the patients carry the KMT2D variant, 5% of the cases carry the KDM6A variant, while for 20% of the cases the etiology remains unknown (6).
Although this syndrome is very rare, it is very challenging for anesthesiologists. In the preoperative assessment we must identify all risks and end-organ involvement. The first and the most important thing for the anesthesiologist is the potential difficult airway management. The patients with KS often (in 60% of the cases) have congenital tooth absence, malocclusion, abnormal dentition, widely spaced teeth, conical incisors, delayed tooth eruption and ectopic upper molars, high arched palate and cleft lip and/ or palate (7). Other features of this syndrome, that are also predictors for difficult airway management, are external ear dysmorphism (dysplasia, enlargement, external rotation, low set or a cup shape) and midface hypoplasia with or without mandibular hypoplasia (8). Equipment for difficult airway management should be available before anesthetic induction, including indirect video-laryngoscopy and different sizes of laryngeal masks. In 2000, Van Haelst et al. described unusual life-threatening complications (not previously reported) in two patients with Kabuki Syndrome that had distal airway abnormalities (9).
Symptoms from central nervous system may include hypotonia (difficulty in the ability to suck, chew and swallow, open mouth in rest), epilepsy, developmental delay (at mild to moderated degree, if there is absence of structural brain abnormalities) and intellectual disability. Ligamentous laxity may be present in up to 90% of the patients with KS on 6 to 14 years of age (10), and this may lead to cervical instability during intubation. According to some studies, epilepsy is present in 5 – 16% in the patients with KS, while others estimate up to 36% (11-13). Since hypotonia is a cardinal feature in patients with KS, anesthesiologists should carefully choose neuromuscular blocking agents. Succinylcholine may be contraindicated due to the risk of malignant hyperthermia, and patients with motor weakness and hypotonia might have increased sensitivity to non-depolarizing neuromuscular blocking agents. There are limited reports of anesthetic care for patients with KS in the literature, and there has been not registered increased sensitivity to non-depolarizing neuromuscular agents (14-16). But this effect should be considered in the cases with associated hypotonia. Also, a larger dose of non-depolarizing neuromuscular agents may be required if the patient is on anti-convulsant therapy.
Congenital heart diseases (CHD) are cardinal features in KS patients. In 2001, Digilio et al. reported in their study with 60 patients congenital heart disease in 58% (35 patients) (17). The three most observed cardiac defects in KS patients are coarctation of aorta (CoA – 23%), atrial septal defect, (ASD – 20%) and ventricular septal defect (VSD – 17%). In 2017, Digilio and the same group of authors reported CHD in 70% of the patients with KMT2D (MLL2) variant (in 19 of 27 patients) and only in one patient with KDM6A gene (18). CoA was again the most common CHD (21%) together with bicuspid aortic valve (21%). Digilio et al. suggest that CoA probably is due to underlying connective tissue diseases. According to these findings, echocardiography must be included in the preoperative evaluation. Attention should be paid to the detection of left-sided obstructive lesions in patients with KMT2D variants, and the right-sided lesions should be considered in patients with KDM6A variants (18).
In patient with pre-existing hypotonia, recurrent pneumonia, chronic aspiration and poor cough effort, it should be considered to use of short acting anesthetic agents, and after long surgical interventions postoperative monitoring for respiratory function is recommended due to increased risk of respiratory failure.
KS patients present urinary system abnormalities in up to 25 – 40% (6,19). Urinary tract malformations include hydronephrosis and ureteral duplication, while renal malformations include horseshoe kidney, renal dysplasia, renal ectopy, renal duplication, and renal insufficiency reported in one case (due to bilateral renal dysplasia at the age of 6 years) (20). Renal malformations are more present in KMT2D variant (21). Also, KS patients undergo surgery for undescended testis.
Endocrine disorders like growth hormone deficiency (22), pituitary hormone deficiency (23), adrenal insufficiency, diabetes insipidus, hypothyroidism and hyperinsulinism with transient neonatal or infantile hypoglycemia (24), have been reported in patients. Cardinal feature present in the patients with KS is short stature as a direct result of GH deficiency. In long surgical interventions, intraoperative glucose monitoring is recommended for detecting and management of hypoglycemia.
In 2010, Teixeira et al. reported a case of a latex allergy in an 11-years-old patient with KS. The patient had a history of allergic reactions after small surgeries for removal of soft tissue lesions, and after preoperative skin allergic tests she was diagnosed with allergy to latex (25).
Emil Bosinci reported case series in 2022, using regional anesthesia in 4 patients with different syndromes, one of which was with KS. No complications were reported preoperatively. The patients were hemodynamically stable while maintaining excellent breathing pattern and without pain and postoperative nausea and vomiting (26).
Conclusion
In perioperative management of patient with Kabuki Syndrome anesthesiologist should take under consideration the possible difficult intubation, neurological (seizure) and musculoskeletal (hypotonia) disorders, cardiac (CoA, ASD, VSD) abnormalities, respiratory problems (recurrent infections), urogenital (hydronephrosis, renal hypoplasia) abnormalities and a latex allergy, as well as the risk of malignant hyperthermia.
References
- Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. 2011 Sep 1 [updated 2022 Sep 15]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. PMID: 21882399.
- Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981; 99: 565-569.
- Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981; 99: 570-573.
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2011, 42.
- Lee JE, Wang C, Xu S, Cho YW, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2013, 2013, 1503.
- Boniel S, Szymańska K, Śmigiel R and Szczałuba K. Kabuki Syndrome—Clinical Review with Molecular Aspects. Genes 2021, 12 (4), 468; https://doi.org/10.3390/genes12040468.
- Porntaveetus T, Abid MF, Theerapanon T, Srichomthong C. Expanding the Oro—Dental and Mutational Spectra of Kabuki Syndrome and Expression of KMT2D and KDM6A in Human Tooth Germs. Int. J. Biol. Sci. 2018; 14.
- Petzold D, Kratzsch E, Opitz C, Tinschert S. The Kabuki syndrome: Four patients with oral abnormalities. Eur. J. Orthod. 2003; 25: 13–19.
- van Haelst MM, Brooks AS, Hoogeboom J, et al. Unexpected life-threatening complications in Kabuki syndrome. Am J Med Genet. 2000; 94: 170-173.
- Upton S, Stadter CS, Landis P, Wulfsberg EA. Speech characteristics in the Kabuki syndrome. Am. J. Med. Genet. Part A 2003; 116: 338–341.
- Verrotti A, Agostinelli S, Cirillo C, D’Egidio C, et al. Long-term outcome of epilepsy in Kabuki syndrome. Seizure 2011; 20: 650–654.
- Kurahashi N, Miyake N, Mizuno S, Koshimizu E, Kurahashi H, et al. Characteristics of epilepsy in patients with Kabuki syndrome with KMT2D mutations. Brain Dev. 2017; 39: 672–677.
- Ogawa A, Yasumoto S, Tomoda Y, Ohfu M, Mitsudome A, Kuroki Y. Favorable seizure outcome in Kabuki make-up syndrome associated with epilepsy. J. Child Neurol. 2003; 18: 549–551.
- Atalay YO, Kaya C, Ustun YB, Sahinoglu AH. Anesthesia management in a patient with Kabuki syndrome. Med Arch. 2014; 68: 359-360.
- Roy D, Das T, Ahmed A, Rudra A, Mitra D. Kabuki syndrome and its anaesthetic management. Indian J Anaesth. 2011; 55: 431-433.
- Casado AI, Ruiz J, Oro J, et al. Anaesthetic management in a case of Kabuki syndrome. Eur J Anaesthesiol. 2004; 21:162-163.
- Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. Congenital heart defects in Kabuki syndrome. Am. J. Med. Genet. 2001; 100: 269–274.
- Digilio MC, Gnazzo M, Lepri F, Dentici ML, Pisaneschi E, Baban A, et al. Congenital heart defects in molecularly proven Kabuki syndrome patients. Am. J. Med. Genet. Part A 2017; 173: 2912–2922.
- Elmitwalli I, Banoub R, Heng R, Tobias JD. Anesthetic care of a child with Kabuki Syndrome. Pediatric Anesthesia and Critical Care Journal 2023;11(1):16-22.
- Ewart-Toland, A, Enns GM, Cox VA, Chandra Mohan G, Rosenthal P, Golabi M. Severe congenital anomalies requiring transplantation in children with Kabuki syndrome. Am. J. Med. Genet. 1998; 80: 362–367.
- Courcet JB, Faivre L, Michot C, Burguet A, et al. Clinical and molecular spectrum of renal malformations in kabuki syndrome. J. Pediatr. 2013; 163: 742–746.
- Ruault V, Corsini C, Duflos C, Akouete S, et al. Growth charts in Kabuki syndrome 1. Am. J. Med. Genet. Part A 2020; 182: 446–453.
- Takagi M, Ishii T, Torii C, Kosaki K, Hasegawa T. A novel mutation in SOX3 polyalanine tract: A case of kabuki syndrome with combined pituitary hormone deficiency harboring double mutations in MLL2 and SOX3. Pituitary 2014; 17: 569–574.
- Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, et al. Congenital hyperinsulinism as the presenting feature of Kabuki syndrome: Clinical and molecular characterization of 10 affected individuals. Genet. Med. 2019; 21: 233–242.
- Teixeira VC, Neves MA, de Castro RA. Latex allergy in a patient with Kabuki syndrome. Case report. Rev Bras Anestesiol. 2010; 60(5): 544–550.
- Bosinci E. Regional anesthesia in surgery of pediatric patients with congenital syndromes – case series. Reg Anesth Pain Med 2022; 47(Suppl 1): A290.