Gjoshevska Dashtevska E1,2, Pandilov S1, Ivanova Cekov M1
1PHI University Clinic for Eye Diseases, Skopje, Republic of North Macedonia
2Faculty of Medicine, University “Ss. Cyril and Methodius”, Skopje, Republic of North Macedonia
DOI: https://www.doi.org/10.55302/MJA2373077gjd
Abstract
Albinism is a large group of hereditary diseases characterized by reduced or completely absent synthesis of melanin in tissues of ectodermal origin. Patients with this pathology have frequent affection of the ocular structures, which are manifested by: reduced visual acuity, transillumination of the iris, hypoplasia of the fovea, nystagmus, strabismus, refractive errors and other. Prevention of UV rays, and early and appropriate management of ophthalmic manifestations is a key link in the treatment of patients with this condition. In this paper we have presented the most significant forms of albinism that are known in science today, as well as the spectrum of ophthalmological changes and their possible treatment.
Key Words: albinism, melanin, nystagmus, ophthalmological manifestations, strabismus.
Introduction
Albinism is a common term for a large group of hereditary diseases, characterized by reduced or completely absent melanin in tissues of ectodermal origin. That is, skin, hair and eyes. The word comes from the Latin term “albus” – white. The biggest feature of this entity is that patients have hypo or depigmentation of the whole body, with prominent gray to white hair. This authentic look is specific to the so-called oculocutaneous albinism (an autosomal recessive condition), in contrast to the ocular albinism, which mainly affects intraocular structures. Albinism is found in the whole population, but also in other species of the animal world. The incidence is different in different nations, with the highest representation among the indigenous population in Panama and Colombia at 6.3 per 1000 inhabitants, and worldwide it ranges between 1:5000 and 1:40,000 (1).
Oculocutaneous Form of Albinism
The oculocutaneous form is based on a mutation of the OCA gene. OCA1A is the most severe type of mutation where melanin synthesis is completely absent, while other variants such as OCA1B, OCA2, OCA3 or OCA4 are relatively deficient in synthesis. OCA2 is the most common mutation in the world. Different mutations are specific to different ethnicities and nations around the globe (2). The OCA group of genes participates in the tyrosine/melanin biochemical cascade. Mutations in this group lead to impaired synthesis of enzymes and membrane proteins that are very important in melanin synthesis. As consequence, no synthesis of this pigment occurs at all, or after it has been synthesized, it cannot be distributed and deposited in the appropriate cellular compartments to exert its effect. Because of these biochemical disorders, the characteristic phenotype of persons with this entity occurs. In other words, there will be a partial or complete affectation of skin pigmentation, hair, iris color, which directly depend on the size and number of melanosomes. The patients in whom residual enzyme activity is present, acquire some pigmentation of the skin and hair (blonde to pale brown), in contrast to those in whom there is an absolute deficiency of enzyme activity, who are presented with completely white skin and hair, as well as light blue eyes with a pronounced red reflex from the fundus (3).
Ocular Albinism
Ocular albinism is a lesser-known entity. The diagnosis is mainly made when nystagmus is present since early childhood, transillumination of the iris with pronounced hypopigmentation of the peripheral part of the retina, generally in males with mild hypopigmentation of the skin and reduced vision. It is mainly an X-linked disease, the symptoms of which begin in the first three to six months of life. There are two forms of the disease: Ocular albinism type 1 (OA type 1) and Ocular albinism type 2 (OA type 2) (4).
The first type, also known as Nettleship-Falls ocular albinism, is an X-linked disease, found almost exclusively in male children. It is the most common form of ocular albinism with a representation of 10% of all albinisms. The incidence is between 1:50,000 to 1:150,000 live births. The disease is due to a mutation of the GPR143 gene from the short arm of the X-chromosome (5). This gene encodes a protein that is extremely important for melanosome transport in pigment cells. The phenotypic characteristics depend on the ethnicity of the affected individual. The diagnosis is made by genetic analysis, as well as by family history. In female carriers of a mutated gene, due to the process of lyonization of the X-chromosome, a Mud-splattered-like change in the fundus can be observed.
The second type, also known as Aland Island ophthalmic disease, or Forsius-Eriksson ocular albinism is a much rarer X-linked disease than the previous one, but with similar clinical manifestations. A characteristic finding is the appearance of protanopia and problems with dark adaptation. Unlike OA1, this condition is not associated to fundus changes in female carriers. The disease is proven genetically by analyzing the gene of interest CACNA1F (6).
Differential Diagnosis
Several systemic diseases and hematological conditions are presented clinically in a similar manner to albinism. In other words, pathophysiological mechanisms that are involved with defective transport and packaging of cellular proteins, clinically manifest in a similar way as albinism. Examples of such diseases are: Hermansky-Pudlak syndrome (autosomal recessive disease); albinoidism (autosomal dominant disease); Waardenburg syndrome (autosomal dominant disease); Chediak-Higashi syndrome (autosomal recessive disease); Griscelli syndrome; Elejalde syndrome and others (2,7-9).
Ophthalmological Manifestations of Albinism
- Transillumination of the Iris
Due to the reduced or completely absent pigmentation in the iris stroma and the posterior pigment epithelium, there are fenestrae through the iris through which a phenomenon of transillumination is observed, in patients with albinism. There are several ways to detect this phenomenon. The slit lamp biomicroscope technique is the most used. A small beam of light is directed through the pupil, during which transillumination defects across the iris surface are displayed in orange, they can be of different shapes and in different distribution and density of arrangement. According to the degree of their presence, as well as the degree of pigment present, schemes with degrees of transillumination from I to IV are used, where IV is the highest degree of transillumination and complete absence of pigment in the iris (10,11). Although this phenomenon occurs in albinism, it is not specific to it, as other ophthalmic conditions are also characterized by the presence of iris transillumination.

Figure 1. Iris transillumination in a patient with albinism.
Source: https://gene.vision/knowledge-base/albinism-for-patients/
- Reduced Vision
One of the main characteristics of patients with albinism is reduced vision. It is still not known in science whether there is a stoppage of previously normal visual development, or if the development is hypoplastic from the beginning in people with this pathology. Visual acuity can range from 20/20 to 20/400, but the most commonly ranges from 20/100 to 20/200. Mainly, vision loss is due to foveolar hypoplasia and disturbed morphological characteristics of photoreceptor cells, especially those in the macular region. High refractive errors are quite rare, but some degree of myopia, hypermetropia and astigmatism is often present. Stereoscopic vision is also affected in patients with a marked degree of albinism, but without affectation of color vision (12).
- Strabismus
Both horizontal and vertical deviations in oculomotor balance may be encountered in patients with albinism. Due to the early development of strabismus, they usually do not show diplopia. Despite the frequent presence of strabismus, amblyopia is rare in these individuals. In contrast, nystagmus is quite a common coexisting manifestation in people with albinism, together with strabismus, and with a large amplitude (12,13).
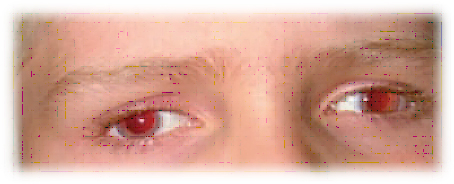
Figure 2. Exotropia with a red fundus reflex present in a child with albinism.
Source: https://www.lecturio.com/concepts/albinism/
- Nystagmus
The most common type of nystagmus is horizontal according to the direction of stretching, and pendular or indeterminate according to its nature. It is usually clinically manifested, especially in children. In different types of albinism, it can appear differently, and thus affects visual acuity. To get better vision, patients with albinism and nystagmus often hold their head in a forced position in order to minimize its effect. Nystagmus in albino individuals causes changes in the retinal image. In those individuals with a marked degree of horizontal nystagmus, extraocular muscle surgery (according to Kastenbaum-Andreson) is a possible option, with modest postoperative results. Retroequatorial insertion of all four straight horizontal muscles is a possible procedure for significant reduction of nystagmus and improvement of the patient’s subjective vision (14,15).
- Photosensitivity and Photoaversion
Patients with albinism often have photophobia, an increased sensitivity to light. The reason for this is generally due to reduced melanin pigment in the cellular structures of the iris and retina. Due to this, there is an inadequate focusing of the light rays, or rather they are scattered and cause dispersed irritation of the photoreceptor cells. In order to improve photosensitivity, patients with this condition are advised to wear hats and protective glasses when in bright environments (16).
- Abnormal Decussation of the Visual Pathways
Each of the mammals has its own characteristic in the percentage of optic nerve fibers that cross at the level of the optic chiasm. For man, that value is about 53%. This ratio is extremely important for achieving adequate stereoscopic vision. In people with albinism, this percentage is disturbed and can be up to 90% crossing. The consequence of this is seen in an inadequate arrangement of the optical fibers at the level of the lateral geniculate and inadequate projection at the level of the visual cortex, which will ultimately have repercussions on stereoscopic vision and in the appearance of strabismus in these individuals (12,17). Monocular visual evoked potentials are important for defining this condition and can be used as a diagnostic tool in non-specific forms of albinism.
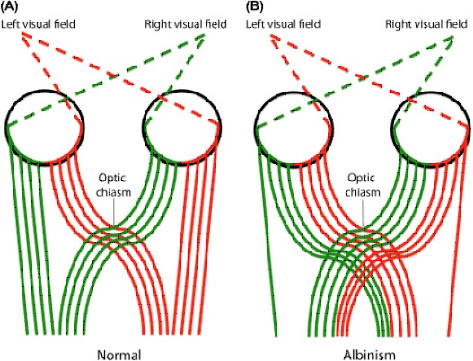
Figure 3. Comparison of chiasm decussation in an individual without and with albinism.
Source: https://www.sciencedirect.com/science/article/abs/pii/B9780128133163000076
- Foveal Hypoplasia and Changes of the Fundus
The overall aspect of the fundus is characteristic in patients with albinism. That is, due to the absence of melanin pigment in the retinal pigment epithelium (RPE), a so-called transparent retina occurs, through which the choroidal blood vessels can be seen, and the scleral reflection, which gives a whitish appearance to the fundus.
Another major feature of fundoscopic examination in these patients is the loss of the foveolar depression due to its hypoplasia. During organogenesis, as a result of disturbed tyrosine metabolism and dysfunction of melanin vacuoles in the RPE, inhibition of signaling occurs, which is important for the development of the fovea and the macular region in general. In the absence of such signal stimuli, the development of this part of the retina lags and remains functionally and morphologically hypoplastic. Foveolar hypoplasia is accompanied by the partial or complete absence of the foveolar avascular zone in these patients, as well as microvascular bridges at the level of the fovea that cross the horizontal meridian of the eye (18).
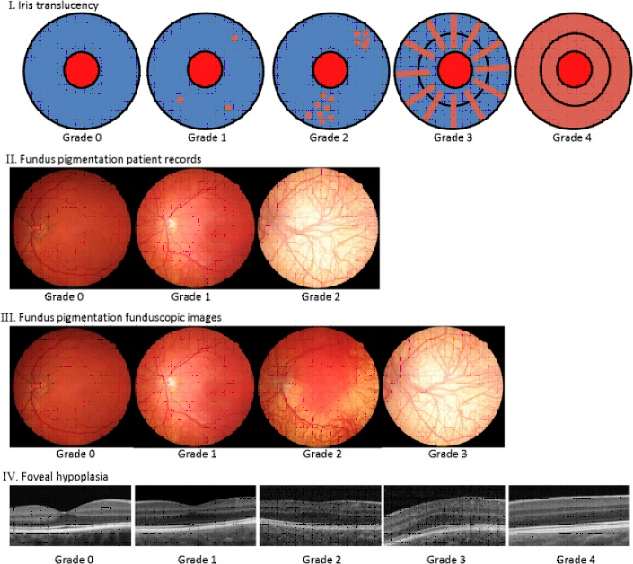
Figure 4. Ocular phenotypic spectrum of albinism.
Source: https://www.sciencedirect.com/science/article/abs/pii/S0161642018305748
Prognosis and Treatment
There is still no causal treatment in patients with albinism. Prevention of exposure to UV rays is extremely important in these individuals because they are at high risk of developing squamous or basal cell skin cancer. Therefore, a much-reduced exposure to the sun, wearing protective clothing, etc. is recommended. Regarding ophthalmological problems, timely recognition of persons with ophthalmological manifestations of the disease is of great importance. Initially, to prevent the development of amblyopia, to correct refractive errors and of course to achieve the best possible visual function. Albino patients who have strabismus or nystagmus may have an improvement if they undergo surgical treatment of the extraocular muscles (2,19).
Several preparations are under investigations that have the potential to find clinical application in the treatment of these patients. One of them is L-DOPA as a replacement therapy because it is an intermediate in this metabolic pathway. Other potential substances are aminoglycosides that would help in bridging non-sense mutations in genetic-biochemical processes at the cellular level. Recently, the drug Nitisinone has been approved by the FDA as an inhibitor of tyrosine degradation in patients with hereditary tyrosinemia. In the future, more studies are expected that would show the possible benefit of this preparation in patients with albinism. The idea is that the reduced degradation of tyrosine will lead to its increased concentration which may induce stimulation of tyrosinase and melanin synthesis. Until now, several animal studies have given good results using this agent (20,21).
Conclusion
Like many other systemic diseases that give ophthalmological manifestations, albinism is one of the entities that presents itself through an ocular affection. The recognition of ocular abnormalities in patients with this pathology is of great importance for timely diagnosis of the condition, all with the aim to provide appropriate treatment and management of the problem, both systemic and local-ophthalmological. Impairment of visual function still remains a major ophthalmological problem in people with ocular or oculocutaneous albinism.
References
- Albahlool AM, Almajed FA, Alwabari ME, et al. Overview on Ocular Manifestations of Albinism- A Review. Journal of Pharmaceutical Research International; 2021:33(40A), pp. 300–308. doi: 10.9734/jpri/2021/v33i40A32248.
- Federico JR, Krishnamurthy K. Albinism. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 30085560.
- Grønskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis. 2007 Nov 2; 2:43. doi: 10.1186/1750-1172-2-43. PMID: 17980020; PMCID: PMC2211462.
- Kubasch AS, Meurer M. Okulokutaner und okulärer Albinismus [Oculocutaneous and ocular albinism]. Hautarzt. 2017 Nov;68(11):867-875. German. doi: 10.1007/s00105-017-4061-x. PMID: 29018889.
- Booth AV, Soldano AC, Levine J, Pomeranz M. X-Linked ocular albinism; Nettleship-Falls ocular albinism. Dermatol Online J. 2008 May 15; 14(5):4. PMID: 18627740.
- Weleber RG, Pillers DA, Powell BR, Hanna CE, Magenis RE, Buist NR. Aland Island eye disease (Forsius-Eriksson syndrome) associated with contiguous deletion syndrome at Xp21. Similarity to incomplete congenital stationary night blindness. Arch Ophthalmol. 1989 Aug; 107(8):1170-9. doi: 10.1001/archopht.1989.01070020236032. PMID: 2667510.
- El-Chemaly S, Young LR. Hermansky-Pudlak Syndrome. Clin Chest Med. 2016 Sep; 37(3):505-11. doi: 10.1016/j.ccm.2016.04.012. Epub 2016 Jun 30. PMID: 27514596; PMCID: PMC4987498.
- Ahmed J, N, Mui RK, Masood S. Waardenburg Syndrome. 2023 Jul 4. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan –. PMID: 32809714.
- Ajitkumar A, Yarrarapu SNS, Ramphul K. Chediak-Higashi Syndrome. 2023 Jul 24. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 29939658.
- Wirtschafter JD, Denslow GT, Shine IB. Quantification of iris translucency in albinism. Arch Ophthalmol. 1973 Oct;90(4):274-7. doi: 10.1001/archopht.1973.01000050276004. PMID: 4746640.
- Kruijt CC, de Wit GC, Bergen AA, Florijn RJ, Schalij-Delfos NE, van Genderen MM. The Phenotypic Spectrum of Albinism. Ophthalmology. 2018 Dec; 125(12):1953-1960. doi: 10.1016/j.ophtha.2018.08.003. Epub 2018 Aug 8. PMID: 30098354.
- Neveu MM, Padhy SK, Ramamurthy S, et al. Ophthalmological Manifestations of Oculocutaneous and Ocular Albinism: Current Perspectives. Clin Ophthalmol. 2022 May 24; 16:1569-1587. doi: 10.2147/OPTH.S329282. PMID: 35637898; PMCID: PMC9148211.
- Hertle RW. Albinism: particular attention to the ocular motor system. Middle East Afr J Ophthalmol. 2013 Jul-Sep; 20(3):248-55. doi: 10.4103/0974-9233.114804. PMID: 24014991; PMCID: PMC3757637.
- Kang NY, Isenberg SJ. Kestenbaum procedure with posterior fixation suture for anomalous head posture in infantile nystagmus. Graefes Arch Clin Exp Ophthalmol. 2009 Jul; 247(7):981-7. doi: 10.1007/s00417-009-1037-2. Epub 2009 Feb 3. PMID: 19189117; PMCID: PMC2686801.
- Dumitrescu AV, Tran J, Pfeifer W, et al. Clinical albinism score, presence of nystagmus and optic nerves defects are correlated with visual outcome in patients with oculocutaneous albinism. Ophthalmic Genet. 2021 Oct;42(5):539-552. doi: 10.1080/13816810.2021.1933544. Epub 2021 Jul 12. PMID: 34251969.
- Hansen TB, Torner-Jordana J, Kessel L. Photosensitivity and filter efficacy in albinism. J Optom. 2023 Jul-Sep; 16(3):214-220. doi: 10.1016/j.optom.2022.07.002. Epub 2022 Aug 24. PMID: 36028395; PMCID: PMC10323186.
- Schmitz B, Krick C, Käsmann-Kellner B. Morphologie des Chiasma opticum bei Albinismus [Morphology of the optic chiasm in albinism]. Ophthalmologe. 2007 Aug; 104(8):662-5. German. doi: 10.1007/s00347-007-1572-3. PMID: 17605013.
- Meyer CH, Lapolice DJ, Freedman SF. Foveal hypoplasia in oculocutaneous albinism demonstrated by optical coherence tomography. Am J Ophthalmol. 2002 Mar; 133(3):409-10. doi: 10.1016/s0002-9394(01)01326-5. PMID: 11860983.
- Moreno-Artero E, Morice-Picard F, Bremond-Gignac D, et al. Management of albinism: French guidelines for diagnosis and care. J Eur Acad Dermatol Venereol. 2021 Jul;35(7):1449-1459. doi: 10.1111/jdv.17275. Epub 2021 May 27. PMID: 34042219.
- Onojafe IF, Adams DR, Simeonov DR, et al. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism. J Clin Invest. 2011 Oct; 121(10):3914-23. doi: 10.1172/JCI59372. PMID: 21968110; PMCID: PMC3223618.
- Mártinez-García M, Montoliu L. Albinism in Europe. J Dermatol. 2013 May; 40(5):319-324. doi: 10.1111/1346-8138.12170. PMID: 23668539.