UDK: 616-056.7:575.224.23
Sokolova R1, Todorovikj L1, Risteski T1, Ljumani Bakiji Lj1, Taleva B1, Mishoska M2
1University Clinic for Pediatric Surgery – Skopje, Republic of North Macedonia
2Clinic Hospital Dr. Trifun Panovski – Bitola, Republic of North Macedonia
Abstract
Bardet-Biedl syndrome (BBS) is a genetic and multisystem disease that affects the genitourinary tract, locomotory system, causes eye anomalies, cognitive disorders and characteristic truncal obesity. It is caused by mutations in certain genes, namely: BBS1 to BBS21 gene. The approach to this disease is multidisciplinary.
Material and Methods: We present a 3-years-old female child who was referred to the Clinic for Pediatric Surgery due to supernumerary toes on both feet. This is postaxial polydactyly. Intrauterine lobulated kidney structure was observed. The child had problems with her peers and avoids socializing with them. The following examinations were performed: cardiological, ophthalmological and genetic. Genetic examinations confirmed the BBS syndrome.
Results: The patient was operated at the clinic for pediatric surgery, the operative and postoperative period were without complications.
Conclusion: BBS is a rare autosomal disease that requires timely detection and appropriate multidisciplinary treatment. This allows complications to be reduced and the child to be included in everyday activities.
Keywords: Bardet-Biedl syndromee; genetic disease; postaxial polydactyly.
Introduction
Bardet-Biedl syndrome (BBS) is a rare multisystem autosomal genetic disorder that affects cells (ciliopathy). Patients with BBS have problems with obesity in which fat tissue being distributed around the abdomen. The patients also suffer in terms of intelligence. Renal, ocular and genital function disorders are known. The first description of this syndrome was done in 1920 by Bardet, and later in 1922 by Biedl.
Truncal obesity is a condition characterized by a disproportionate distribution of adipose tissue around the abdomen, and less commonly on the chest and extremities. This phenomenon is known as the “apple-shaped body”. The child’s weight is usually normal at birth, but a noticeable change occurs quickly in the first year of life. Patients with BBS suffer from non-insulin-dependent diabetes in 45% of the cases. Obesity in these patients is manifested in 72%-92% of the cases (1).
These patients may have changes in the cardiovascular system, such as defects in the heart muscle, valve stenosis, that result in arrhythmias.
Retinal changes in this syndrome may appear around the 7th and 8th year of life as “nail blindness”, which progresses to varying degrees. Affected individuals first lose peripheral vision and look directly at a frontal point. Over time, retinal degeneration occurs. Additional eye changes in affected individuals include strabismus, cataracts and glaucoma (2).
Patients with BBS have abnormalities of hands and feet. Postaxial polydactyly, syndactyly (8% between the second and third fingers), brachydactyly (46%), clinodactyly, and sandal gap between the first and second fingers have been described. Approximately, these changes occur in about 63% – 81% of the patients. Polydactyly can be present in all four limbs (21%), only in the upper limbs (8%), or only in the lower limbs (21%) (3).
Other cardinal abnormalities in BBS are the following: reduced or poor gonadal function; Hypogonadism is present in 59% of the patients. In boys, it is characterized by undescended testicles, micropenis, recessive penis and reduced testicular volume. Undescended testicles are a high risk for testicular cancer. Affected girls may have genital and urinary abnormalities, such as: underdeveloped fallopian tubes, underdeveloped ovaries and uterus, irregular menstrual cycles and polycystic ovaries. Hydronephrosis involves frequent bacterial infections that can result in pyelonephritis, chronic kidney damage and dialysis, up to and including kidney transplantation (4).
Cognitive impairment is a characteristic of this syndrome. It is present in 60%-66% of the patients (5). It manifests as learning difficulties, dysfunction of brain development, neurological changes such as poor coordination, large and small motor tremors, inability to play with friends, etc.
Certain individuals with BBS also have liver problems, such as dilation or stricture of the bile ducts, which is present in 30% of the patients. Digestive problems such as Hirschsprung’s disease, ciliary disorders, inflammatory changes, stenoses and other anatomical anomalies of the intestine are rare (6). The prognosis of this disease depends on the degree of kidney damage and progressive vision loss.
Incidence
BBS affects both males and females equally. The distribution of this disease is not homogeneous. It occurs in 1 in 120,000 to 1 in 160,000 births in the population of North America and Europe (7). In Sweden, it occurs in 1 in 160,000 births. A high frequency of this syndrome occurs in the Bedouins of Kuwait, 1 in 13,500 births (8).
Cause of BBS
BBS can be caused by mutations in more than 20 different genes. It is an autosomal recessive disease. There are several gene mutations that lead to the development of BBS:
BBS1 gene, BBS2, BBS3, BBS4, BBS5 BBS6, BBS7, BBS8, BBS9, BBS10 to BBS 21 gene.
The risk of the disease occurring in a child of both parents carrying the gene is 25% for each pregnancy. The risk of the gene occurring in a child of both parents carrying the gene is 50% for each pregnancy (9).
Diagnosis
Diagnosis is based on clinical examinations and associated clinical manifestations. Some patients remain undiagnosed for years. Genetic testing may aid in diagnosis. Definitive diagnosis requires family history, clinical signs, neurological testing, ophthalmologic evaluation, audiometry, ECG and echocardiography, abdominal ultrasonography and renal function testing. Laboratory testing includes complete blood count, glucose tolerance test, liver enzymes, hormonal status and genetic testing. Cardinal diagnostic features include the following: retinal degeneration, obesity, postaxial polydactyly, renal anomalies, neurologic intellectual disability, hypogonadism and genitourinary abnormalities. Secondary features of BBS include musculoskeletal abnormalities, hearing loss, cutaneous dermatoses. The presence of four of the cardinal signs or three cardinal and two secondary signs is sufficient for a diagnosis of BBS. Genetic testing confirms the diagnosis.
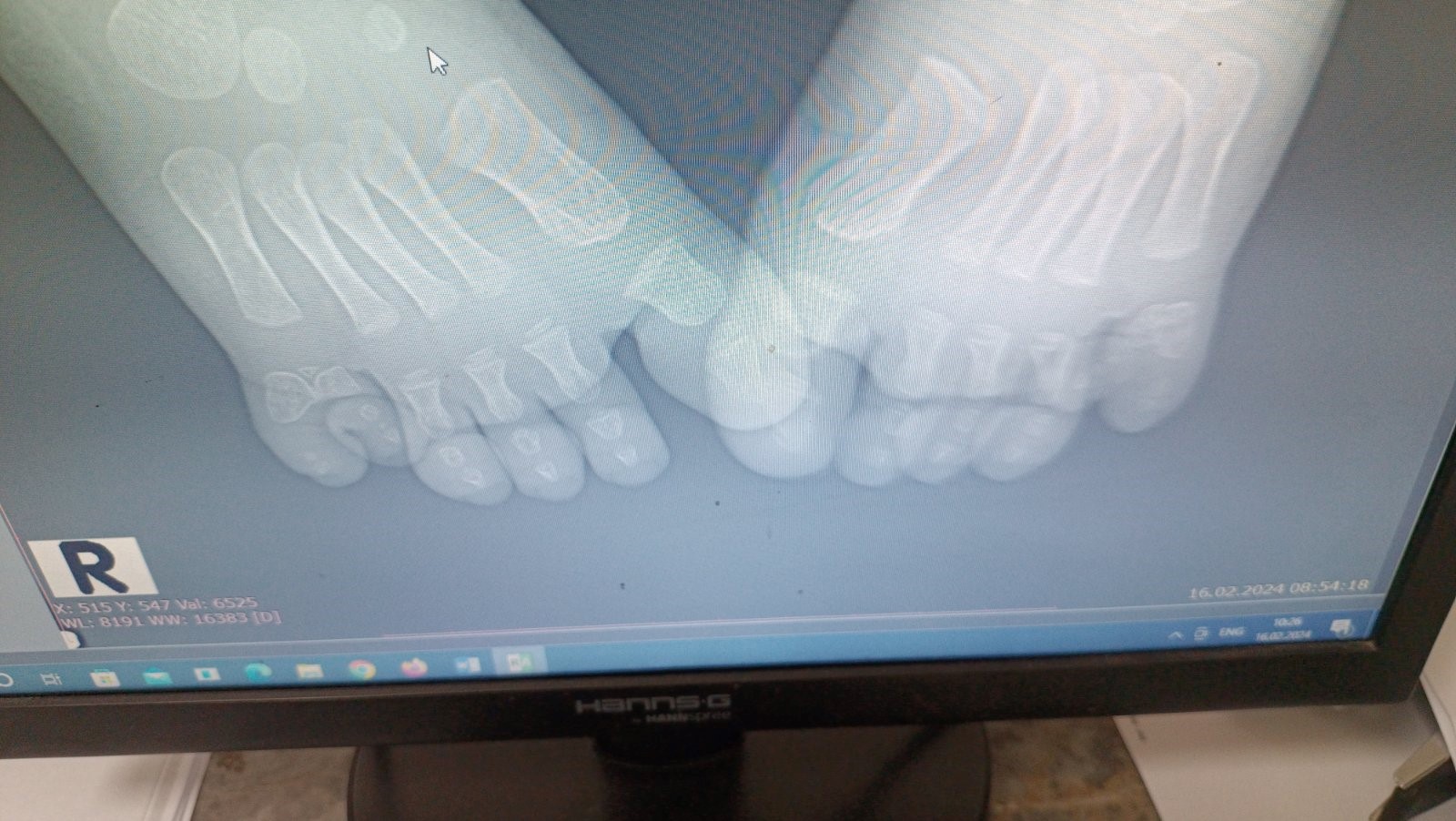
Picture 1. Native radiography of both feet of a child with BBS.
Differential Diagnosis
Laurence-Moon syndrome (LMS) – LMS is a rare autosomal recessive disorder characterized by visual degeneration and pituitary dysfunction. Patients have neurological problems including loss of motor control, loss of peripheral nerve function and intellectual disability.
Alstrom syndrome – A rare autosomal recessive disorder characterized by hearing and visual abnormalities, obesity and progressive renal dysfunction. This syndrome also includes cardiac muscle dysfunction and intellectual disability.
Meckel syndrome – This syndrome is in a group of rare autosomal disorders characterized by protrusion of part of the brain, predominantly encephalocele, multiple renal cysts, polydactyly, hepatic fibrosis and genital abnormalities.
McKusick-Kaufman syndrome (MKKS) – It is an autosomal recessive genetic disorder characterized by postaxial polydactyly, congenital heart defects, hydrometrocolpos, gastrointestinal abnormalities, genitourinary anomalies and renal anomalies.
Biemond syndrome – This syndrome is an autosomal recessive genetic disorder. It is characterized by the absence of tissue in the eyes (iris coloboma), intellectual disability, obesity and genitourinary abnormalities.
Treatment for patients with BBS – The treatment is multidisciplinary and depends on the symptoms of the affected individuals. Some of the abnormalities can be corrected with surgical treatment (genitourinary abnormalities, congenital heart defects, polydactyly, kidney transplantation and ophthalmological corrections). Obesity should be treated with specific diet programs, hobbies with athletics, and consultation with a nutritionist for the prevention of obesity. In 2022, the Food and Drug Administration (FDA) approved the drug setmelanotide for the treatment of chronic obesity.
Discussion
BBS is historically known as Laurence-Moon-Biedl-Bardet syndrome and was described by scientists as the first case of this syndrome. Cardinal signs of BBS are truncal obesity, intellectual disability, renal anomalies, polydactyly, retinal degeneration and hypogonadism.
Case report
A 3-years-old female child was admitted to the Clinic for Pediatric Surgery due to polydactyly of both feet. From the family history, the mother has hypothyroidism and one grandmother died of pancreatic cancer. The change is congenital. The child was examined at the Clinic for Pediatric Diseases due to changes in both kidneys that were seen intrauterine. This is the second child from a second regular and properly controlled pregnancy. Born in the 38th week of gestation with a birth weight of 3,050 grams and a birth length of 48cm. Apgar was 9/10. Vaccination started regularly and established regularly according to the calendar. Examined in the nephrology outpatient clinic of the Clinic for Children’s Diseases due to hyperechogenic kidneys with a granular structure. The remaining findings of abdominal ultrasonography were with normal parameters. The heart findings were normal, clear tones without systolic murmur and rhythmic action. The lung findings were within normal limits, mediastinal structures were properly positioned. Laboratory analyses: blood count, glycemia ionogram, hepatogram, proteinogram, AST and ALT, urea and creatinine were within normal limits.
Molecular genetic studies are compatible with the clinical diagnosis of autosomal recessive type BBS type 12 in our patient. Segregation analysis of variants confirmed the biallelic status in our patient with c8656>C inherited from the father (DNA ID 29101) and c 1658T> C from the mother (DNA ID 29100). These data were obtained from the Institute of Human Genetics – University Hospital Cologne.
Retinal echo was normal. Fundus examination with indirect biomicroscopy, PNO (optic nerve papilla) at the level of the retina with clear borders and normal color. Blood vessels were normal and Macula lutea (ML) was normal, without signs of retinopathy.
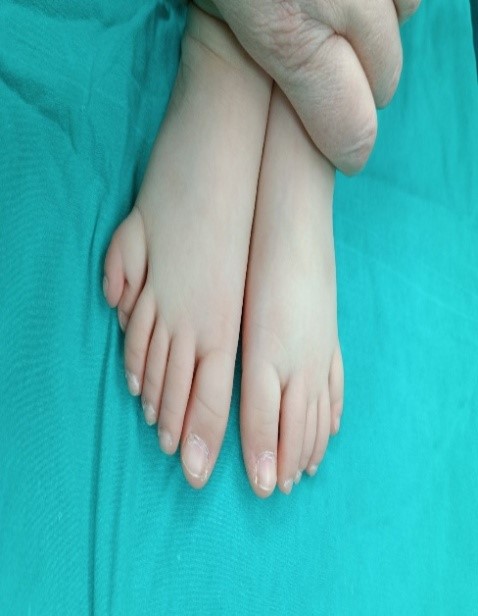
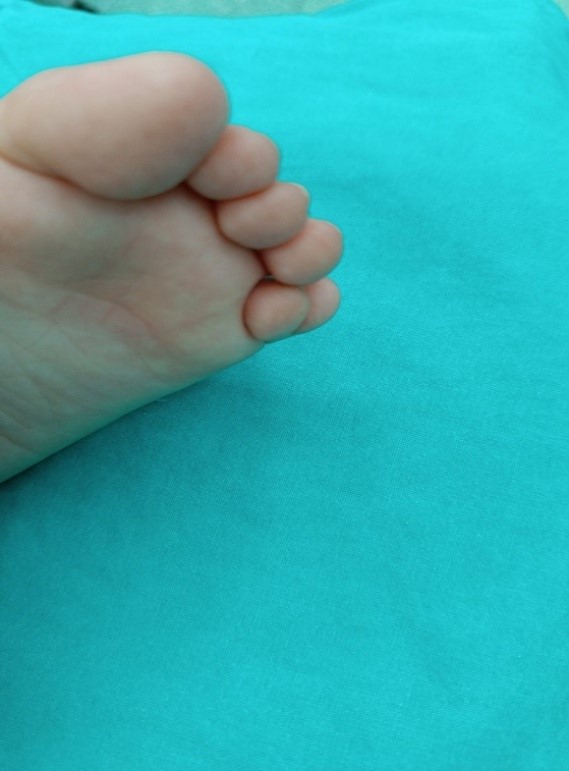
Picture 2. Before surgery. Picture3. Before surgery.
A female child was admitted to the Clinic for Pediatric Surgery for surgical correction of axial polydactyly of both feet. X-ray in addition to postaxial polydactyly, Dg. Polidactilia Axial Ispedis bill. Based on the performed anesthesiologic examinations, surgical intervention was permitted. Laboratory examinations were within normal limits. The surgical intervention was performed under endotracheal anesthesia. Excision of the toes of both feet that were outside the axis was performed. On both feet, the accessory toes were located between the 4th and 5th toes. Local plastic surgery of the foot followed without the use of skin transplantation. Dressing was done with antibiotic gauze. Waking up was orderly. The postoperative course was orderly. Wounds were healed per primam. Finger movements were orderly.
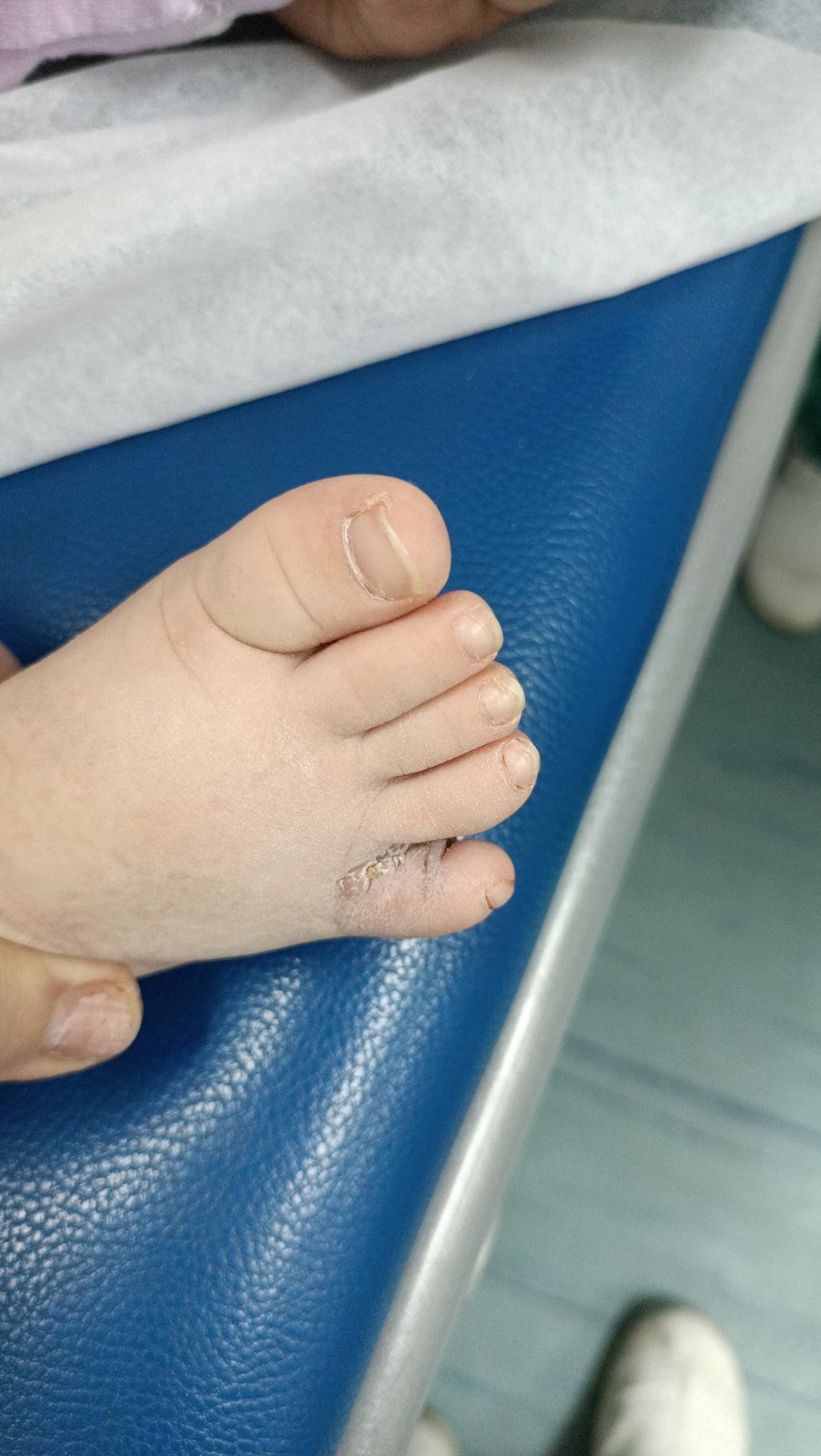
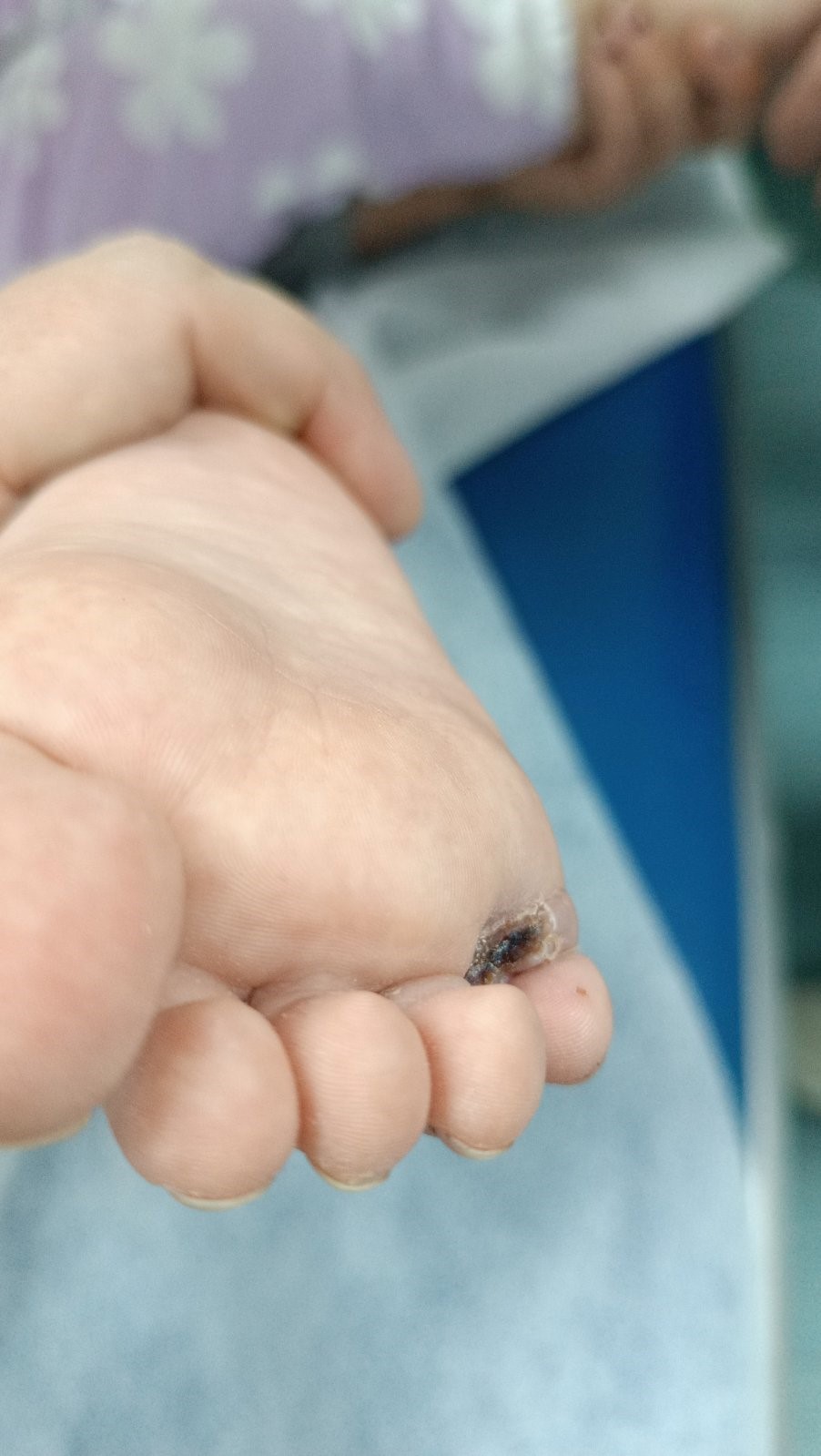
Picture 4. Postoperative local finding. Picture 5. Postoperative local finding.
Gene therapy research is an advance in BMS. Certain BMS mutations can be targeted with oligonucleotides. In vivo, potential therapeutic effects have also been demonstrated with fibroblasts. Over the past 10 to 15 years, studies have shown that loss of cilia at the cellular level or their dysfunction are numerous causes of BMS.
Pomeroy et al. observed an increase in sleep time that may be beneficial in alleviating obesity (10). Seo et al. published a study in 2009 on intravenous melanocortin receptor agonists for weight reduction and feeding (11). Mujahid describes pituitary abnormalities in 19.5% of the female population with BBS (12). Certain individuals with BBS may develop abnormalities in kidney structure and function. Renal anomalies include fetal lobulation of tissue, small kidneys, ectopic or duplex kidneys, tubular or interstitial nephritis, glomerulosclerosis, polyuria and others (13).
Setmelanotide is a melanocortin-4 receptor (MC4R) agonist that reduces weight and hunger. The Food and Drug Administration approved the treatment for chronic obesity in adults and pediatric patients of 6 years of age and older in 2022.
Ganawa et al. describe the success of using a GLP-1 agonist to reduce body mass index (BMI) (14). A mega-analysis was published by Niederlova et al. (15).
They analyzed the largest cohort study of BBS patients with a total of 889 patients, pooling data from 85 studies focusing on gene-phenotype correlations. Geno-phenotype correlations were used for disease severity. Patients with mutations and presumed loss of function had a higher syndromic score than the rest.
Prognosis
Depending on the symptoms, the survival rate varies. Moor and colleagues describe a median survival of about 63 years (16). Myocardial infarction results in mortality from 40 to 54 years. O’Dea describes in his study renal abnormalities from 19-60 years, then embolism with thrombosis from 32 to 34 years, gastrointestinal hemorrhages at 45 years (17).
Conclusion
BBS is a rare genetic disease due to ciliopathy. Due to the involvement of multiple systems, the approach to these patients is multidisciplinary. The diagnosis can be made even intrauterine. A multidisciplinary approach is mandatory in this disease due to multiorgan dysfunction. The role of cellular biology is important for the development of specific therapy.
| Patient: | _________ (female) | Sample received | 23.02.20 |
| Date of birth: | 03.02.2021 | Request received | 23.02.20 |
| Ident. Sample Number: | 29099 | Sample processed | 18.06.20 |
| Sample type: | DNA | (date of sampling: unknown) | Payment
Science |
Reason for testing:
Diagnosis/assumption of autosomal recessive Bardet – Biedl Syndrome
Requested analisys
NGS analysis based on an exome for known genes for renal ciliopathy, using the Agilent SureSelectXT HS Human All Exon V8. Bioinformatical analysis of 520 genes related to renal pathology of genomic DNA. Alignment of the sequence with human hg38 reference genome. Analysis of coding gene regions and 20 bp of neighboring, non-coding regulatory sequence. The sequencing was done on the Illumina NovaSeq 6000 platform.
Clinical details and previous findings
Patient with bilateral hyperechoic kidneys with a status of previous polydactyly
Results
Filtering for potential complex heterozygosity in 520 genes related to kidney pathology produced a result of only two different heterozygotic BBS12 variants in the patient.
References:
- Forsythe E. Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013., 21 (1)..8 -13.doi..10.1038/ejhg.2013.115.
- Priya S. Nampoothiri S. Sen P. Sripriya S. Bardet-Biedl sundrome.. genetics, molecular pathophystology, and disease management. Indian J Ophthalmol.2016.,64(9).. 620-627 doi.. 10 4103/0301-4738
- Beales PL. Elcioglu N. Woolf AS, Parker D. New criteria for improved diagnosis of Bardet-Biedl sybdrome results of a population survey. J Med Genet.1999.36(6) .,437-446.doi..101136/jmg 36.6
- Zacchia M, Zacchia E, Zona E,et al.Renal phenotype in Bardet-Biedl syndrome.. a combined defect of urinary concetracion and dilution is associated with defective urinary AQP2 and UMOD excretion.Am/Physiol Renal Physiol 2016..311(4)..F686-F694.doi..10.1152
- Baker K. Northam GB, Chong WK, Nanks T, Beales P. Neocortocal and hippocampal volume loss in a human ciliopathy.. a guantitative MRI studi in Bardet-Biedl syndrome. Am/Med Genet. 2011..1..155(!)..1-8 doi.. 1002/ajkmg.a.{
- Branfield Day L, Quammie C, Heon E, et al. Liver anomalies as a phenotype parameter of Bardet-Biedl syndrome, cli.Genet,2016.,89(4)..507-509 doi 10.1111/cge.12684 {PubMed}.
- Beales Pl. Warner Am, Hitman GA, thakker R, flinter FA. Bardet-Biedl syndrome.. a molecular and phenotypic study of 18families. J Med Genet.1997.,34(2)..92-98.doi 10.1136/jmg,34.2.92
- BaragTI. Teebi As. High incidence of Bardet-Biedl syndrome among the Bedoin.Clin Genet 1989.,36(6)463-464.doi 10.1111/j.1399-0004.1989
- Harville HM, Held S. Diaz-Font A, et al.Identification of 11 novel mutation in eight BBS genes by high-resolution homozygosity mapping. J Med Genet..2010.,47(4) ..262-267.doi. 10.1136/jmg.2009.071365
- Pomepoy J, Van Worner JJ, Meilahn JR, Maki T, Haws RM. Sleep and physical activity patterns in adult and children with BBS. Orphanet J Rare Dis.2021.,16(1). Doi 10.1186/s13023-021-01911-4
- Seo S, Guo DF, Bugge K. Morgan DA, Rahmouni K, Sheffield VC.Reguirement of BBS proteins for leptin receptor signalling Hum Mol Genet. 20098., 1897) 1323-1331. Doi.. 10 1093/hmg/ddp031
- Mujahid S. Hunt KF. Cheah YS, et al.The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population.JClinEndocrinol Metah.2018..103(5)..1834-1841.doi..10.1210/jc,2017-01459
- Moore Sl, Green JS. Fan Y. et al. Clinical and genetic epidemiology of BBS in newfoundland a 22 year prospective, population-bassed, cohord study. Am J. Med Genet A. 2005.,132(4) ..352-360 doi..10.1002/ajmg.a.30406
- Ganama S, Santhosh SH, Parry L, Syed AA. Weight loss with glucagon –like peptide-1 receptor agonist in BBS. Clin. Obes. 2022. Doi..10.1111/cob12546
- Niederlova V, Modrak M, Huranova M, Stepanek O, Meta analisys of genotype- phenotype associations in BBs uncovers differnces among causative genes., Hum Mutat.2019,40(11).,2068-2087.doi.,10.1002/humu.2386
- Marchese E, Ruoppolo M, Perna A, Capasso G, Zacchia M. Exploring key challenges of undesending the pathogenesis of kidney disease in Bardet-Biedl syndrome. Kidney Int Rep. 2020.,S24680249203401.doi..10.1016/j.ekir2020.06.017
- O.Dea D. Partfrey PS, Harnett JD, Hefferston D, Cramer BC, GreenJ. The importance of renal imparment in the natural history of BBS. AmJ Kidney Dis. 1996.,27(6).,776-783.doi 10.1016/So272-6386(96)90513-2