UDK: 615.015.2
Andonovska B1
1University Clinic for Traumatology, Orthopedic Diseases, Anesthesia, Reanimation, Intensive Care and Emergency Centre, University “Ss. Cyril and Methodius”, Skopje, Republic of North Macedonia
Abstract
Drug interactions can be described as the pharmacological influence of one drug on another drug, when administered in combination. Drug interaction can cause an increased or decreased effect of the drug, but it can also lead to a toxic effect. In daily practice in operating rooms and intensive care units, anesthesiologists routinely combine drugs. Interactions are usually divided according to the mechanism of occurrence and the most frequent are pharmacokinetic and pharmacodynamic. In pharmacokinetic interactions, the interactions are at the level of absorption, distribution, metabolism, or elimination processes. Such interactions are predictable, but their extent cannot be predicted. Pharmacodynamic interactions refer to antagonistic or synergistic action between drugs. It remains a challenge to teach clinicians how to combine these drugs in order to achieve and maintain optimal anesthetic and vital conditions, while minimizing side effects. A good understanding and knowledge of drug interactions can improve the ability to titrate multiple drugs more effectively.
Key Words: anesthesiology, drugs, intensive care, interaction.
- Introduction
The concept of drug combination has been known and used since ancient times to treat diseases and reduce suffering of people. In the process of anesthesia or treatment in intensive care units, doctors have to reach therapeutic decisions which routinely include administration of several drugs, and almost every day they face anesthetic-drug interactions or drug-drug interactions. Speaking with the language of statistics, between 46% and 90% of the patients in intensive care units are exposed to potential drug-drug interaction, and this percentage is twice higher compared to patients in other units (1).
Critically ill patients and anesthesiology patients differ from other hospital patients and outpatients for several reasons: very often these patients have a serious degree of injury or are in an advanced stage of a disease, in advanced age, with often present impeded or altered absorption, metabolism disorders, reduced kidney and liver function and polypharmacy, and hence these patients are more vulnerable to drug-drug interactions. On the other hand, since these patients are closely monitored, some adverse events from drug-drug interactions are more acceptable than in patients who are not subjected to intensive care or are in the operating theater because vigilant monitoring enables effective and timely risk management and safe treatment (2).
- Definition and positive effects of drug interactions
A drug interaction occurs when one or more drugs influence on pharmacokinetics and/or pharmacodynamics of one or several other drugs.
In anesthesiology, a drug interaction is defined as an influence of one anesthetic over the behavior of another anesthetic.
Combination of drugs can contribute to enhancement, diminishing or onset of a new effect. New effects can be therapeutic, that is, the idea of combination – interaction of drugs is creation of positive effects by their mutual application, such as:
- Production of synergistic effect on the target,
- Diminishing the necessary drug dosage until it produces the same effect,
- Toxicity reduction,
- Decrease of incidence and number of side effects,
- Minimization or delay in development of resistance.
However, a drug interaction can also cause side effects. In intensive care patients, about 16% of all side effects are caused by drug-drug interactions and are associated with a longer hospital stay, higher morbidity and mortality and increased hospital costs (3).
- Types of interactions
Examination and modeling of drug interactions is one of the priorities of modern pharmacology (4).
Due to concomitant administration, drug interactions can appear across different stages of drug action (4). Therefore, drugs can be subjected to physical and chemical interaction prior to administration and absorption or across the processes of different pharmacological phases: pharmacokinetics, with potential influence on absorption, distribution, metabolism and elimination, or pharmacodynamics, with alteration of pharmacological effects (4,5).
These different mechanisms of interactions can generate negative results, with increased toxicity or interference without the therapeutic effect, but also can be used as a strategy for useful interactions, with a potential to increase the pharmacological effect and reduce toxicity.
3.1.Pharmaceutical Interactions
Pharmaceutical interactions occur prior to drug administration, and they are changes in the physical-chemical structure of one drug affected bythe action of another drug when combined in the same solution whether in a bag, syringe, or an infusion system. This type of interaction provides information about drug stability and compatibility that is examined with chromatography (6).
Whenever two or more drugs are mixed for anesthesia or other treatment, or when those drugs share the same infusion line, the question about their compatibility must be raised.
The combination of remifentanil and propofol is very common in some medical institutions. Literature data report that stability of this mixture combining these two drugs depends on time, the proportion of remifentanil -propofol and the recipient. Thus, it can be concluded that remifentanil can be combined with propofol, but the above mentioned conditions have to be taken into consideration (7).
The mechanism responsible for pain at the site of propofol injection is mediated by the kallikrein-kinin pathway and bradykinin production, a process that can be inhibited by lidocaine. But is it possible to combine propofol and lidocaine in order to prevent such pain? Are these compounds compatible? Literature reports that adding lidocaine to propofol increases the diameter of the lipid molecules, which makes this mix physically and chemically unstable and poses a potential risk of pulmonary embolism.
Figure 1 illustrates the pharmaceutical interactions of the most commonly used drugs in anesthesia.
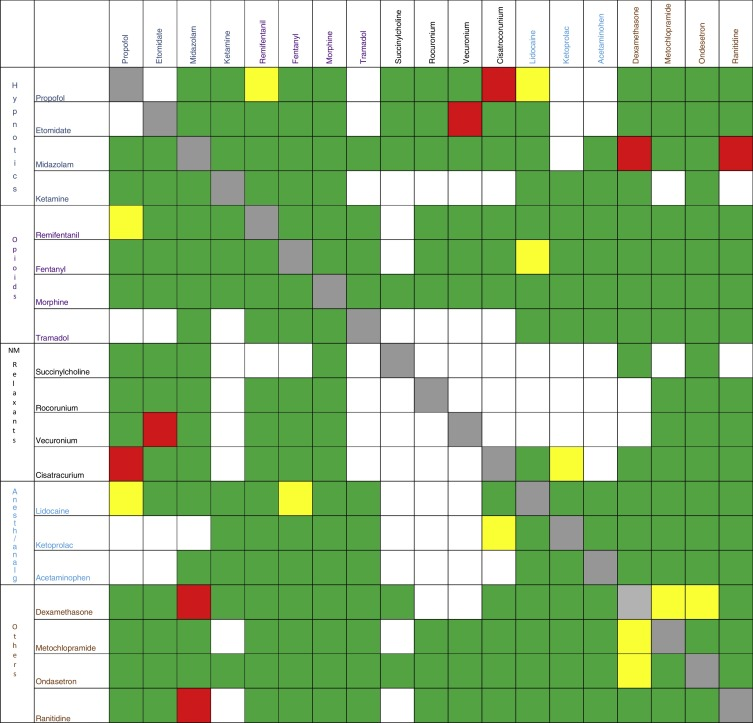
Figure 1. Pharmaceutical interactions of the most commonly used drugs in anesthesia.
Red box: incompatible; green box: compatible; yellow box: inconclusive [9].
3.2.Pharmacokinetic Interaction
Pharmacokinetics is officially defined as the measurement and interpretation of the changes in drug concentration in one or more parts in the organism in a unit of time, that is, it is mainly focused on drug plasma concentration.
Co-administration of drugs, herbal medicines and food can induce changes in the pharmacological bioavailability – drug concentration, described as pharmacokinetic interaction. During pharmacokinetic interaction, the concomitant administration of one drug alters the absorption, distribution, metabolism and excretion of the other drug. Due to individual differences in these processes, pharmacokinetic interactions can be expected, but their intensity cannot be anticipated (10).
3.2.1Absorption
Absorption is defined as the passage of a drug from the site of administration into plasma.
The mainroutes of drug administration are as follows:
- oral,
- sublingual,
- rectal,
- application on other epithelial surfaces (e.g.,skin, cornea, vagina, and nasal mucosa),
- inhalation,
- injection.
Orally administered drugs are absorbed in the gastrointestinal tract at different sites with or without the help of different carriers, with passive or active transfer and at rate determined by the degree of ionization and liposolubility. The largest number of interactions at the level of absorption affects the absorption rate and, to a certain degree, the extent of absorption. Absorption rate is not important if the amount of the absorbed drug is not significantly changed. Delayed absorption can be clinically important in drugs with short life of half-elimination and when rapid maximum plasma concentration of the drugs is required. Very often, interaction at the level of absorption results in absorption decrease, not increase, and can be avoided if the application of two drugs is done in an interval of two to three hours.
The most significant mechanisms of a drug interaction at the level of absorption that would create conditions for changes in the absorption are: pH of the gastrointestinal tract, disorders in the intestinal microflora, motility and viscosity of the mucus, as well as pathological conditions of inflammatory, metabolic, neurological or autoimmune character.
Some drugs manifest instability in acidic media, such as penicillin G, erythromycin and digoxin, when variations in gastric pH influence the rate of drug degradation and bioavailability. In cases like this, pretreatment with omeprazole or an increase in pH will enhance the absorption (11).
Concurrent use of drugs and food containing bivalent metal ions can also change the pharmacological availability due to formation of insoluble, inactive complexes. The co-administration of norfloxacin with milk or yoghurt will reduce its bioavailability by 50%; this also refers to tetracyclines (11).
For example, sucralfate works by forming a barrier over the gastrointestinal mucosa, thus protecting it from ulcer development. It acts so as to reduce the bioavailability of fluoroquinolones by forming stable chelate complexes between aluminium in its molecule and fluoroquinolones and hence, absorption is reduced.
The rate of gastric emptying and intestinal motility define the rate and extent of the drug absorption (12).
Metoclopramide is a prokinetic that stimulates the serotonin 5-HT4 receptors, antagonizes the presynaptic inhibition of muscarinic receptors, and blocks the dopamine D2 receptors. Thus, the release of acetylcholine leads to an increase in the intragastric pressure, which is responsible for acceleration of gastric emptying. When administered simultaneously with other drugs, metoclopramide tends to increase the absorption of other compounds because it exposes them more quickly to the intestinal absorption area (13).
On the other hand, opioids such as morphine, due to their action on opioid receptors in the myenteric plexus, and anticholinergic drugs such as atropine, by blocking the muscarinic receptors influence on reduction of motility and delayed gastric emptying. Both mechanisms tend to reduce the absorption of the concomitantly administered drugs (14).
Also, in conditions of hypovolemia or heart failure, transport through splanchnic circulation is reduced resulting in reduced drug resorption.
In case of intramuscular or subcutaneous administration, the absorption depends on the local blood flow, on drug ionization or lipid solubility to a great extent. For example, the administration of combined local anesthetic and vasoconstrictor will lead to reduced absorption of the local anesthetic and prolonged action. Transdermal route can be used for drugs that are highly soluble in lipids (e.g., fentanyl) where delayed absorption in the end creates permanent concentrations in the blood.
When anesthetic drugs are administered intravenously, problems with the absorption are avoided to a great extent. By using volatile anesthetics, the absorption can be under the influence of the gradient of the anesthetics’ partial pressures in the alveoli and circulation, that is, ventilation-perfusion ratio or membrane pathology, but also of the ventilator parameters.
3.2.2 Interactions during Distribution
Distribution is the movement of a drug in the body. Once absorbed, the drug is distributed through systemic circulation to extra- or intracellular space until it reaches its target organ. If a drug from the beginning is in systemic circulation and then it is distributed to the other parts of the organism, then it is a one-compartment model. If a drug is first bound to a parenchymal organ and then is distributed to target sites, then it represents a two-compartment model, and achieving the full effect of the drug will be prolonged. With reference to volatile anesthetics and some intravenous drugs, one can talk of a three-compartment model, which is basically a depot of the drug in the organism (usually it is a fat tissue releasing a certain drug amount that goes to the effectors), and results in a prolonged recovery from the action of the anesthetic. Drug distribution is influenced by regional circulation, pH of the environment, drug chemical characteristics, drug lipid solubility, plasma protein binding and tissue binding. Regarding drug distribution, it is noteworthy that in this phase interaction can also happen, and one drug can change the distribution of another drug. The most common interaction in the phase of distribution is a result of the competitive relationship for the binding site of albumins or tissue proteins. The consequence is displacement of one drug and replacing it with another. Replacement of a drug at the binding site will quickly lead to an increase in concentration of the released drug, and this situation is followed by a compensatory increased metabolism and elimination of the free drug. This can result in a decrease of the total concentration of the displaced drug, but the free drug concentration remains similar to that before introduction of the other displacing drug. These interactions are usually clinically insignificant except in case of disordered metabolism or elimination, and clinical consequences include:
- toxicity due to transient increase in concentration of free drug before the steadystate is reached, or
- when the displacing drug additionally reduces the elimination of the first, then the free drug concentration is increased not only acutely but also chronically, which can lead to severe intoxication (15).
Secondly, drugs that diminish the heart minute volume can reduce perfusion of tissues included in redistribution of other drugs, thereby changing their extent of distribution. For example, reduced doses of propofol in presence of esmolol have been determined probably as a result of changes in the distribution.
3.2.3 Interaction at the Level of Metabolism
The most important clinical interaction includes the action of one drug on the metabolism of another drug. Metabolism is the process of biochemical drug modification for easier excretion from the organism. Liver plays the central role in drug metabolism, but this process can also be done by the kidneys, lungs, intestines, skin and placenta.
Drug metabolism is divided into two phases. Reactions of the first phase are oxidation, hydrolysis and reduction, whereas conjugation with glucuronic and sulphate acid are reactions of the second phase. Reactions of the first phase take place in the liver mediated by cytochrome P450 and CYP enzymes as the most important enzymes in drug metabolism. Drug interactions can happen due to induction or inhibition of cytochrome P450, which consequently affects changes in the metabolism of one drug. Therapeutic effects of enzyme induction or inhibition depend on pharmacological characteristics of a drug or its metabolites. If one drug is metabolized with cytochrome P450, and another drug inhibits or reduces enzyme activity, then plasma concentration of the first drug remains high for a longer period and their inactivation is delayed. The result of such interaction is prolonged therapeutic response that consequently increases the risk of toxicity. Inhibitory drugs have a competitive feature of binding to cytochrome P450 creating a stable complex that prevents binding of other drugs to cytochrome. Midazolam is a drug that is commonly used as a preoperative sedative, with variable behavior and influence on the metabolism of other drugs because of the inhibition of cytochrome P450 3A4. In comparison with fentanyl, midazolam reduces norfentanyl production by almost 95%. With reference to propofol, metabolism disorder has been reported and its increase in the blood concentration byaround 25%. Such changes in drug metabolism will probably cause events such as depression, hypotension and bradycardia. These events can likely be prevented if these interactions are taken into account. Other drugs that are frequently used and have influence on this enzyme as inhibitors are: dexamethasone, prednisolone, ketamine, antidepressants and alfentanil, amiodarone, alopurinol, ciprofloxacin, diltiazem, isoniazid, intraconazole, metronidazole, omeprazole, oral contraceptives, etc. Theinhibition of cytochrome P450 enzyme depends on the applied dose of the inhibitory drug, and the inhibition starts when minimally inhibitory drug concentrationin the liver is achieved.
If, on the other hand, drugs cause an increased enzyme activity, then concentration of the first drug rapidly reduces and fast inactivation happens. Drugs that are enzyme inducers are rifampicin, barbiturates, phenytoin and carbamazepine. Enzyme induction results ina reduced pharmacological effect of the drug except in cases when metabolites are pharmacologically active. The process of induction depends on the applied drug dose – inducer (16).
3.2.4 Interactions that Affect Excretion
Drugs can be eliminated from the body by excretion (for e.g., kidney excretion of sugammadex, and kidney and biliary excretion of rocuronium), biotransformation (for e.g., liver metabolism of propofol), or spontaneous degradation (for e.g., Hofmann degradation of cisatracurium).
The process of excretion consists of final elimination of compounds in unchanged form or bio-transformed into highly polar metabolites. This process can occur via different routes such as sweat, tears, breast milk, bile, saliva, urine or in a gaseous form via the lungs.
Biliary elimination has to be emphasized since many hydrophilic drug conjugates are concentrated in the bile and are transported into the small intestine and are again regenerated into the active form of the drug that can be reabsorbed, hence, the cycle repeats (enterohepatic circulation). As a result of this process, a reservoir of the recirculating drug is created, which can prolong the action of the drug.
Kidney excretion is the principal way responsible for the elimination of majority of drugs and metabolites. However, any change in the drug binding to proteins and consequently change in its filtration, inhibition of the active tubular secretion, changes in the urine flow and changes in urine pH, are mechanisms by which one drug affects the rate of kidney excretion of another drug (15).
In this context, drug ionization can vary according to urine pH; acidic drugs display increased excretion in basic urine and basic drugs in acidic urine. This characteristic can be used as antidote strategy in cases of intoxication with phenobarbital, for e.g., when sodium bicarbonate is administered for urine alkalization and phenobarbital elimination (17).
3.3Pharmacodynamic Interactions
Drug pharmacodynamic interactions occur when the pharmacological effect of one drug has been altered by the effect of another drug in a combined regimen at the site of its action or by physiological mechanisms. There are three different types of interaction: additive, supra-additive (synergism) or infra-additive (antagonism).Additive interactions are present when two or several drugs with similar mechanisms of action are administered, and the effect of such combination is equal to the expected one by summing their effects. Additive behavior is typical for hypotonic agents and their concomitant use results in an increased number of adverse events because each individual action is not being enhanced, but, on contrary, it is replaced. This type of interaction can be seen when administering sevoflurane and propofol simultaneously.
Synergistic interactions appear when the combination of drugs results in a significantly higher effect in comparison to the expected one for the addition of the effects. This type of interaction is ideal for practicing anesthesia because the necessary dose of two drugs concomitantly administered is lower in comparison to individual separate drug doses. In other words, the effect of the drug is improved when both drugs are present.
The necessity of building synergistic models in anesthesiology imposes inclusion of drugs that act on different receptors and that such drugs would cover the complete hypnosis spectrum in case of hypnotic agents or complete analgesic spectrum in case of opioids. At this point, the risk of using drugs with poorly differentiated pharmacodynamic profiles to reach the expected goal has to be emphasized. For example, when speaking of dexmedetomidine, an α2 agonist receptor, as a drug with increased interest for application in anesthesiology and intensive care, it does not meet all characteristics of a hypnotic agent, nor those of an analgesic agent; it is unable to cover the complete spectrum of hypnosis of analgesia. What does that mean then? In case of requiring a deep hypnosis, its poor efficacy requires the addition of another hypnotic agent. The same principle is valid for analgesia.
Antagonistic or inhibitory interactions occur when the combination of drugs results in a lower effect than the excepted one by the summation of effects (18,19).
Types of pharmacodynamic interactions and examples are shown in Table 1.
Table 1. Types of pharmacodynamic drug interactions.
| Interaction | Combined effect (C) compared to summing individual drug effect | Examples of favorable outcome | Examples of unfavorable outcome |
|
SYNERGISM |
C>A+B |
Aminoglycosides + Penicillin
Penicillin is a bactericidal antibiotic that kills bacterial cell wall that also enhances aminoglycoside transport into the cell and its bactericidal effect. |
Barbiturate + opioid agents
Both groups cause depression of the CNS acting on different target (tissues), but cause similar effects that are thus enhanced: sedation and respiratory depression. |
|
ADDITIVE |
C=A+B |
Aspirin + Acetaminophen
Acetaminophen has no anti-inflammatory effect but enhances the antipyretic and analgesic effect of Aspirin. |
Macrolides + Quinolones
Both antibiotic groups might cause heart arrhythmia. |
|
ANTAGONISM |
C<A+B |
Opioids + Naloxone
Naloxone as an opioid receptor agent blocks the effects of opioids in case of acute poisoning, i.e., respiratory depression. |
Warfarin + VitaminK
Added vitamin K impairs anticoagulation that is being maintained by Warfarin. This can lead to insufficient or unsuccessful anticoagulant therapy which shortens INR. |
3.4.Thermodynamic Interaction
Thermodynamic interaction is an indicator of a drug-receptor interaction at a molecular level and shows the processes of affinity and intrinsic activity. Affinity and intrinsic activity are two separate steps in creating receptors’ pharmacological response under the influence of drugs. Affinity is the strength of a drug to bind to its receptor. Intrinsic activity is the ability of a drug to activate the receptor to which it is bound, but some drugs differ in their potentials for activation depending on the tissue they are located although the receptors are same. When two drugs act on the same receptor and are simultaneously administered, the affinity coefficient (Ki) shall be considered in order to establish which of the two drugs will preferably bind to the receptor regardless of the internal activity. The concept of affinity is based on the concept of power, when the concentrations of both drugs are high, the saturation of the receptors by the higher affinity drug will prevail. A more potent drug usually has high affinity for the receptors, and so it binds to a significant number of receptors even if administered in a lower concentration. Using several drugs that act on the same receptor is not advised and is outdated; it would be like using both captopril and enalapril for blocking angiotensin. These recommendations cannot be overlooked even in the case of opioids administered simultaneously when their mechanism of analgesia prevails over the same receptor. Therefore, the concept of affinity is probably the most important concept when multimodal anesthesia is applied. Based on the concept of multimodal anesthesia, it would be ideal to block our target from different points or regimes. Figure 2 illustrates the concept of multimodal anesthesia.

Figure 2. Multimodal anesthesia concept. Left: multimodal anesthesia, different drugs requiring different receptors (remifentanil R, propofol P and lidocaine L). Right: unimodal anesthesia, several drugs requiring one single receptor (remifentanil, fentanyl and methadone), the drug binding to the receptor would be that with the highest affinity [9].
Based on severity, drug interactions can be classified as:
- Major – the interaction may be life-threatening or can cause permanent impairment;
- Moderate –deterioration of a patient’s clinical condition can be developed, or a patient’s prolonged hospital stay that may require additional care;
- Minor – the interaction can be unpleasant, but not medically harmful.
Table 2 presents the pair of drugs, degree of severity as a consequence of their interaction.
Table 2. The most common prescribed pair of drugs with drug-drug interactions, severity and mechanism of interaction (20).
| Drugs | Severity | Mechanism of interaction |
| Fentanyl – Midazolam | Major | Increases depression of CNS. |
| Dopamine -Noradrenaline | Moderate | Increases blood pressure and heart rate. |
| Adrenaline -Noradrenaline | Moderate | Increases blood pressure and heart rate. |
| Fentanyl – Fluconazole | Moderate | Increases the serum concentration of Fentanyl. |
| Acetylsalicylic acid -Enoxaparin | Moderate | Moderately increases the anticoagulant effect. |
| Midazolam Morphine | Moderate | Increases depression of CNS. |
| Adrenaline – Dopamine 1 | Moderate | Increases depression of CNS depression. |
| Fluconazole – Midazolam | Moderate | (May) Moderately increase(s) the serum concentration of Midazolam. |
| N-acetyl cysteine NTG | Minor | Increases vasodilation effect of NTG. |
| Clarithromycin – Midazolam | Major | Increases the serum concentration of Midazolam. |
| Furosemide – Morphine | Moderate | Reduces the therapeutic effect of Furosemide. |
- Software programs for drug interactions
Drug-drug interactions are a significant problem. It is difficult to remember all drugs and their possible clinically important interactions. Software programs can help in avoiding or reducing the harmful effects of drug interactions. There are many programs, but the most significant are: ePocrates Rx, Tarascon, Pharmacopoeia Deluxe, the mobile PDR, Mobile Micromedex, Lexi-Interact, iFacts, the Medical Letter’s Handbook of Adverse Drug Interactions, Mosby’s Drug Consult Software and Clinical Pharmacology on Hand.
These programs have to meet the following basic characteristics: sensitivity, specificity, positive predictive value and negative predictive value.
Sensitivity is defined as the ability of the software program to recognize interaction drug pairs that are clinically important.
Specificity is defined as the ability of the software program to ignore interaction drug pairs that are not clinically important.
Positive predictive value is the probability that interaction recognized by the software program is defined as a drug interaction clinically important.
Negative predictive value is the probability when the software program ignores a drug interaction; it is defined as not clinically important.
Studies have assessed iFacts and Lexi-Interact software programs to be the most competent and comprehensive. Software programs can be of great help to health professionals, but they cannot be the unique or single information about drug interactions. Drug software programs have many limitations, and the main drawback is the inability for individualization because an interaction between two drugs does not depend solely on those two drugs, but also on a patient’s condition and surrounding factors. It is necessary to develop new programs.
Lexi-Interact is a complete program for retrieval and analysis of interactions between drugs, food and alcohol. When entering the drug data (keywords), interactions can be anticipated, and each possible interaction is defined with a level of clinical importance. Levels of clinical importance are assigned large letters from the alphabet – A, B, C, D and X (Table 3) [21,22].
Table 3. Categorization of drug interactions in Lexi-Comp®Online program.
| Grade/level of clinical significance | Procedures | Description |
| A | No known interactions. | Data show neither pharmacodynamic nor pharmacokinetic interactions between the chosen drugs. |
| B | No intervention is necessary during treatment. | Data show potential interaction between the chosen drugs. There is little or no evidence of the existence of a clinically significant interaction as a result of concurrent use of drugs. |
| C | Necessary follow-up of a patient throughout the treatment period. | Data show that the chosen drugs can have clinically significant interaction; the benefits of simultaneous use of these drugs overweigh the risk. Monitoring patients is indispensable in order to recognize on time the possible negative events. A small number of patients may require dosage adjustment. |
| D | Eventualrequirement for therapy adjustment. | Data show that both drugs can have a clinically significant interaction. Each patient has to be assessed whether the concomitant use of the drugs overweigh the risk. Procedures are to be undertaken in order to estimate the use and minimize toxicity resulting from simultaneous application of those drugs. Procedures include patient’s monitoring, dosage adjustment, selection of alternative drugs. |
| X | Avoiding the combination. | Data show that both drugs can have a clinically significant interaction. The risk associated with concomitant use of these drugs is higher than beneficial effect in majority of cases. The combination of these drugs is mainly contraindicated. |
- Conclusion
The critically ill population and anesthesiology patients are a particularly vulnerable category of patients. A combination of drugs is necessary for their successful treatment. A good knowledge of the administered drugs and their interactions, as well as continuous monitoring of patients will result in successful treatment.
References:
- Zheng W.Y., Richardson L.C., Li L., Day R.O., Westbrook J.I., Baysari M.T. Drug-drug interactions and their harmful effects in hospitalised patients: a systematic review and meta-analysis. Eur J Clin Pharmacol. 2018;74(1):15–27.
- Papadopoulos J., Smithburger P.L. Common drug interactions leading to adverse drug events in the intensive care unit: management and pharmacokinetic considerations. Crit Care Med. 2010;38(6 Suppl): S126–35.
- Reis A.M., Cassiani S.H. Adverse drug events in an intensive care unit of a university hospital. Eur J Clin Pharmacol. 2011;67(6):625–32.
- Li Y., Meng Q., Yang M., Liu D., Hou X., Tang L., Bi H. (2019). Current trends in drug metabolism and pharmacokinetics. Acta Pharmaceutica Sinica B, 9(6), 1113-1144.
- Niu J., Straubinger R.M., Mager D.E. (2019). Pharmacodynamic drug–drug interactions. Clinical Pharmacology & Therapeutics, 105(6), 1395-1406.
- Marsilio N.R, Silva D.D, Bueno D. (2016). Drug incompatibilities in the adult intensive care unit of a university hospital. Revista Brasileira de Terapia Intensiva, 28, 147-153.
- Menéndez Gómez J.M., Betancourt Tafur L.A., Cifuentes Quintero I.F., Vega Figueroa S.P., Murillo Serna A.M., Ramos Gutiérrez A. Manual single infusion of combined remifentanil and propofol for anesthesia during laparoscopic gynecology procedures: a case series. Rev Esp Anestesiol Reanim, 57 (2010), 220-223.
- Jalota L., Kalira V., George E., Shi Y., Hornuss C., Radke O. Prevention of pain on injection of propofol: systematic review and meta-analysis. BMJ, 342 (2011), pp. d1110.
- Tafur-Betancourt L.A. The hidden world of drug interactions in anesthesia. Colombian Journal of Anesthesiology (2017). 45(3), 216-223.
- Akbulut M., Urun Y. (2020). Onco-cardiology: Drugdrug interactions of antineoplastic and cardiovascular drugs. Critical Reviews in Oncology/Hematology, 145, 102822.
- Abuhelwa A.Y., Williams D.B., Upton R.N., Foster D.J. (2017). Food, gastrointestinal pH and models of oral drug absorption. European Journal of Pharmaceutics and Biopharmaceutics, 112, 234-248.
- Corrie K., Hardman J.G. (2011). Mechanisms of drug interactions: Pharmacodynamics and pharmacokinetics. Anaesthesia & Intensive Care Medicine, 12(4), 156-159.
- Shakhatreh M., Malik Z., Parkman H.P. (2019). Metoclopramide for the treatment of diabetic gastroparesis. Expert Review of Gastroenterology & Hepatology, 13(8), 711-721.
- Feng X.Q., Zhu L.L., Zhou Q. (2017). Opioid analgesics-related pharmacokinetic drug interactions:From the perspectives of evidence based on randomized controlled trials and clinical risk management. Journal of Pain Research, 10, 1225.
- Rang P.H., Riter J.M., Flower R.J., Henderson G. Farmakologija.2019 ISBN 978-86-7478-501-0.
- Neves L.M.B. et al. Drug Interactions Pharmacology: A Narrative Review. American Journal of Pharmacology and Toxicology 2022, Volume 17: 27.36.
- Garza A.Z., Park S.B., Kocz R. (2023). Drug elimination. StatPearls Publishing; 2023.
- Nieuwenhuijs D.J.F., Olofsen E., Romberg R.R., Sarton E., Ward D., Engbers D., et al. Response surface modeling of remifentanil–propofol interaction on cardiorespiratory control and bispectral index. Anesthesiology, 98 (2003), 312-322.
- Manyam S.C., Gupta D.K., Johnson K.B., et al. Opioid-volatile anesthetic synergy: a response surface model with remifentanil and sevoflurane as prototypes. Anesthesiology, 105 (2006), 267-278.
- Dagden M.S., Gulen D., Ceylan I. Evaluation of potential drug-drug interactions in intensive care unit. European Review for Medical and Pharmacological Sciences 2021; 25: 5801-5806.
- Barrons R. Evaluation of personal digital assistant software for drug interactions. Am J Health-Syst Pharm 2004;61:380-385.
- Lexi-Comp Online. Avaliable at: http://online.lexi.com/crlsq/servlet/crlonline. Accessed August 31, 2012.